Smart-Seq pipeline
Smart-seq and Smart-Seq2 are popular protocols for single-cell RNA-sequencing. With these protocols, 96-well plates are used with individual cells placed in each well.
To assess the quality of the reads, our pipeline employs FastQC and MultiQC, with checks including distributions of GC-content and levels of adapter contamination. Subsequently, reads are aligned to the appropriate reference genome using STAR, and protein-coding features are quantified with featureCounts program.
10x pipeline
10x genomics has developed several protocols for single-cell RNA-sequencing. Unlike Smart-Seq, 10x protocols are droplet-based, and use unique molecular identifiers (UMIs) to avoid counting an RNA fragment more than once. Typically, 10x data contains significantly more cells, sequenced at lower depth, compared to smart-seq.
Our pipeline uses FastQC and MultiQC to assess the quality of the raw fastq files before alignment and feature quantification with 10x cellranger software.
Single nuclei 10x
Single-nucleus RNA-Seq is gaining popularity as an alternative way to obtain transcriptome profiles of tissues where extracting intact cells is challenging and from frozen samples. Characterized by a lower feature count and a high intronic read content, snRNA-Seq is able to dissect cellular diversity providing similar resolution for cell type detection to scRNA-Seq.
Similar to standard 10X, our workflow for single-nuclei droplet data involves running fastQC and multiQC to check quality of input data and CellRanger for alignment and quantification. A comprehensive QC inspection is required to confirm viability of experimental data, as the intrinsic low input material for single-nuclei 10X may condition moving forward or not in the analysis.
Optimal number of clusters
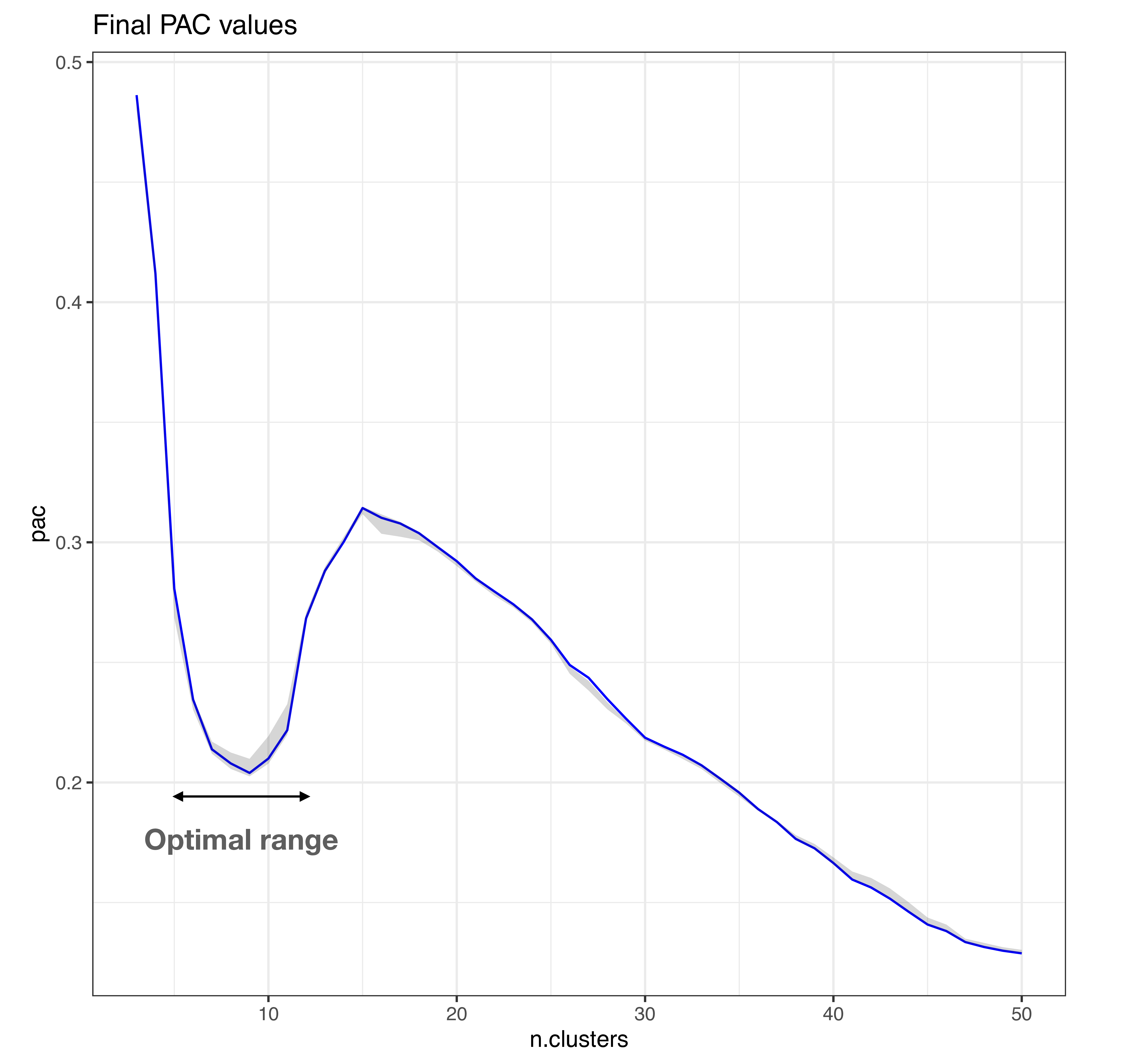
To identify and characterise groups of samples or cells (e.g. for single cell sequencing, corresponding to the same cell type, or for bulk sequencing, to the same developmental stage, etc), we use unsupervised learning approaches i.e. clustering. To determine the optimal number of clusters in a data-driven and unbiased way, our pipeline is based on a measure called proportion of ambiguous clusterings (PAC). Briefly, we repeatedly subsample cells/samples and perform clustering on the subset. By measuring how often each pair of samples cluster together over all subsamples, the robustness of the clustering can be determined. We compare this robustness across different numbers of clusters, and thus identify the number of clusters that leads to the least ambiguous partitioning of samples into groups. This number can then be adjusted based on biological feedback.
In the figure below, the local minima in a PAC landscape corresponds to the optimal range for number of clusters in the data.