Chromatin immunoprecipitation sequencing (ChIP-Seq) is used to analyse DNA-protein interactions. It infers the positions in a reference genome that are bound by the protein of interest. The list of proteins that can be analysed with ChIP-Seq includes transcription factors and repressors, histones and histone modifications, chromatin remodelling proteins, etc.
A standard ChIP-Seq protocol includes:
1. Cross-linking of DNA and the protein of choice, for example, by using formaldehyde treatment that creates reversible links. A protocol without the cross-linking step is known as native ChIP.
2. Chromatin fragmentation is undertaken to make it more accessible to antibodies.
3. Chromatin immunoprecipitation (ChIP) is performed using an antibody specific to the protein of interest. This step allows us capture and elute the DNA fragments bound by the protein.
4. DNA recovery and purification is followed by removal of cross-links and sequencing library preparation.
5. High-throughput sequencing.
A typical ChIP-Seq data analysis pipeline is provided in the figure below.
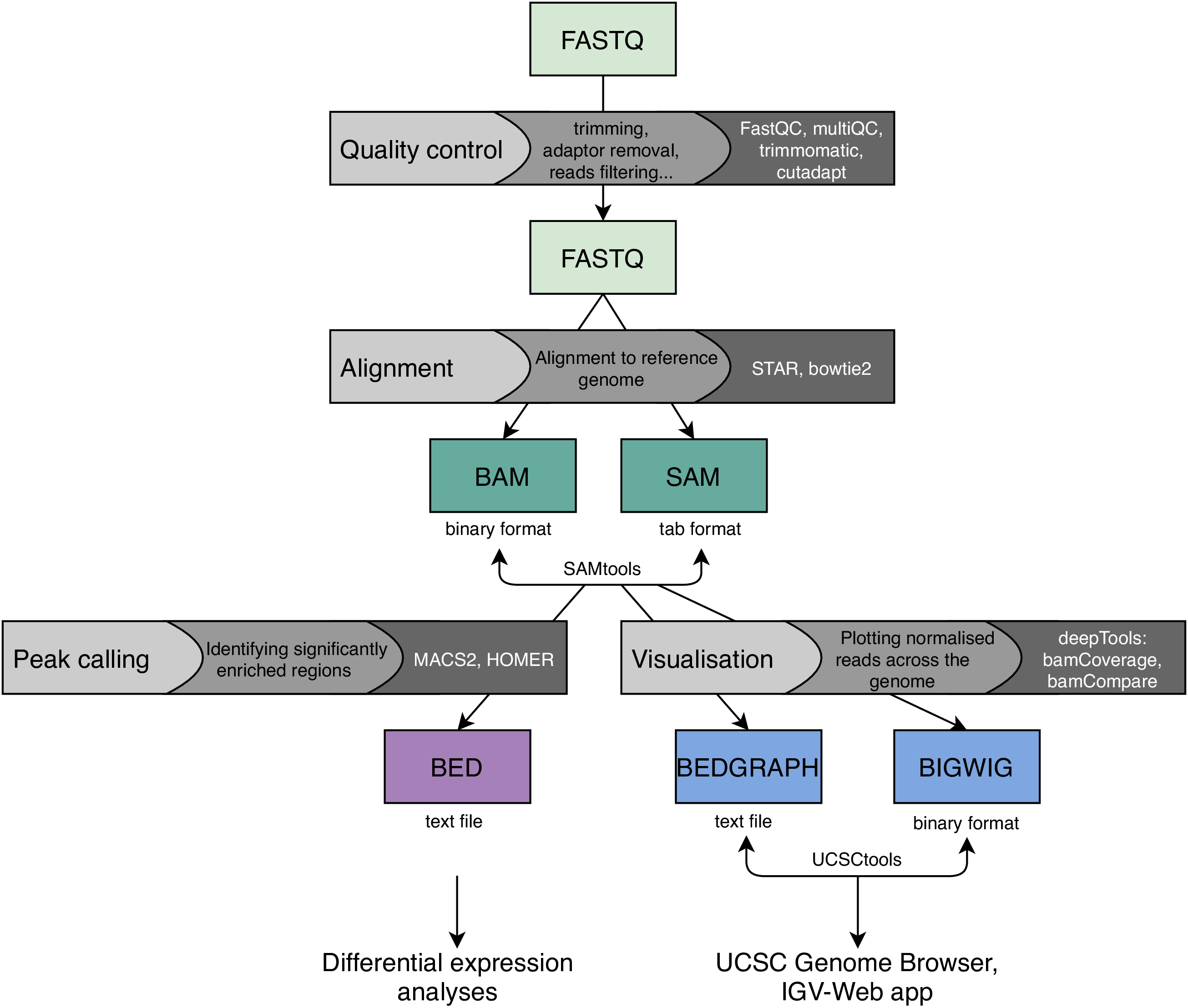
As with all types of high-throughput sequencing, raw data must go through rigid quality control first. Adapter contamination and PCR duplication are one of the typical technical problems that can be dealt with at this stage. Next, ChIP-Seq data is mapped to a reference genome using standard genomic aligners, such as STAR or Bowtie2.
In order to identify the genomic regions that are significantly enriched with the protein of interest, peak calling is performed. It is essential to compare the enrichment in a sample of interest to an input control, which is a sample that has been cross-linked with the protein of choice but has not been immuno-precipitated. Peak calling can be performed to identify either narrow peaks, corresponding to transcriptional factors, or broad peaks, which are more typical of histones that are bound to DNA across wide regions. Both types of peak calling can be performed with MACS2 or HOMER. The identified peaks can be further analysed using differential expression analyses across different samples and sample types. These are typically performed using in-house scripts written in R.
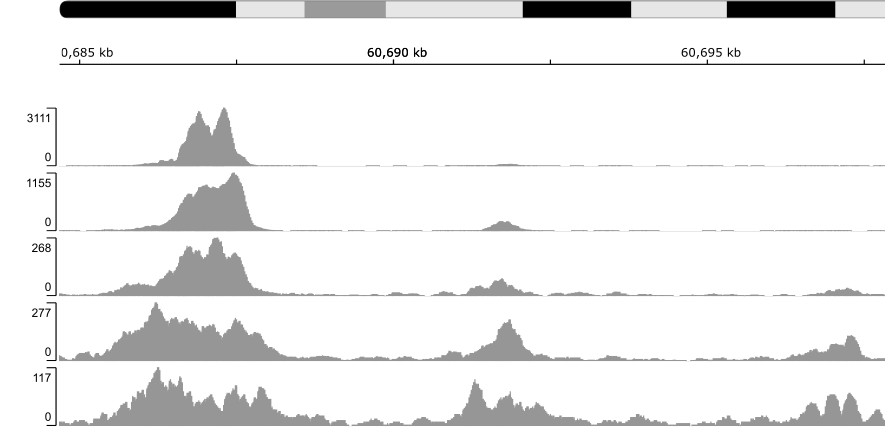
Finally, read coverage across the whole genome can be visualised and compared between samples using tracks. The bamCoverage and bamCompare tools from the deepTools suite can be applied to create either sequencing-depth normalised tracks for individual samples, or input-normalised tracks for sample-input pairs. The most common file format for tracks is BIGWIG that can be viewed through various genome browsers, for example, the UCSC Genome Browser. The figure below depicts varying H3K4me3 enrichment across wide genomic interval in five samples.