Bisulfite sequencing (BS) is a technique used to analyse patterns of DNA methylation. As DNA polymerase used in polymerase chain reaction (PCR) during a standard NGS protocol does not distinguish between methylated and unmethylated cytosines, DNA methylation patterns are difficult to analyse experimentally. In BS, prior to sequencing, DNA is treated with bisulfite, which converts unmethylated cytosine residues into uracil while methylated cytosines remain unchanged. Subsequent PCR amplification converts these uracils into thymines. The introduced changes in the DNA sequence thus directly relate to the methylation status of individual cytosines.
In whole-genome (WG) BS, the entire genome is represented in the library. To achieve sufficient read coverage of each cytosine across the genome, high sequencing depth is required. Thus, WGBS is an expensive technique, especially for studies of organisms with a large genome size. More recently, reduced representation (RR) BS technique was developed, which focuses only on methylation-prone regions of the genome. In RRBS, DNA is fragmented using a restriction enzyme (usually MspI), which cuts at 5’-CCGG-3’ sites, present mostly at genes, promoters and CpG islands, producing a sequencing library that is enriched with these regions.
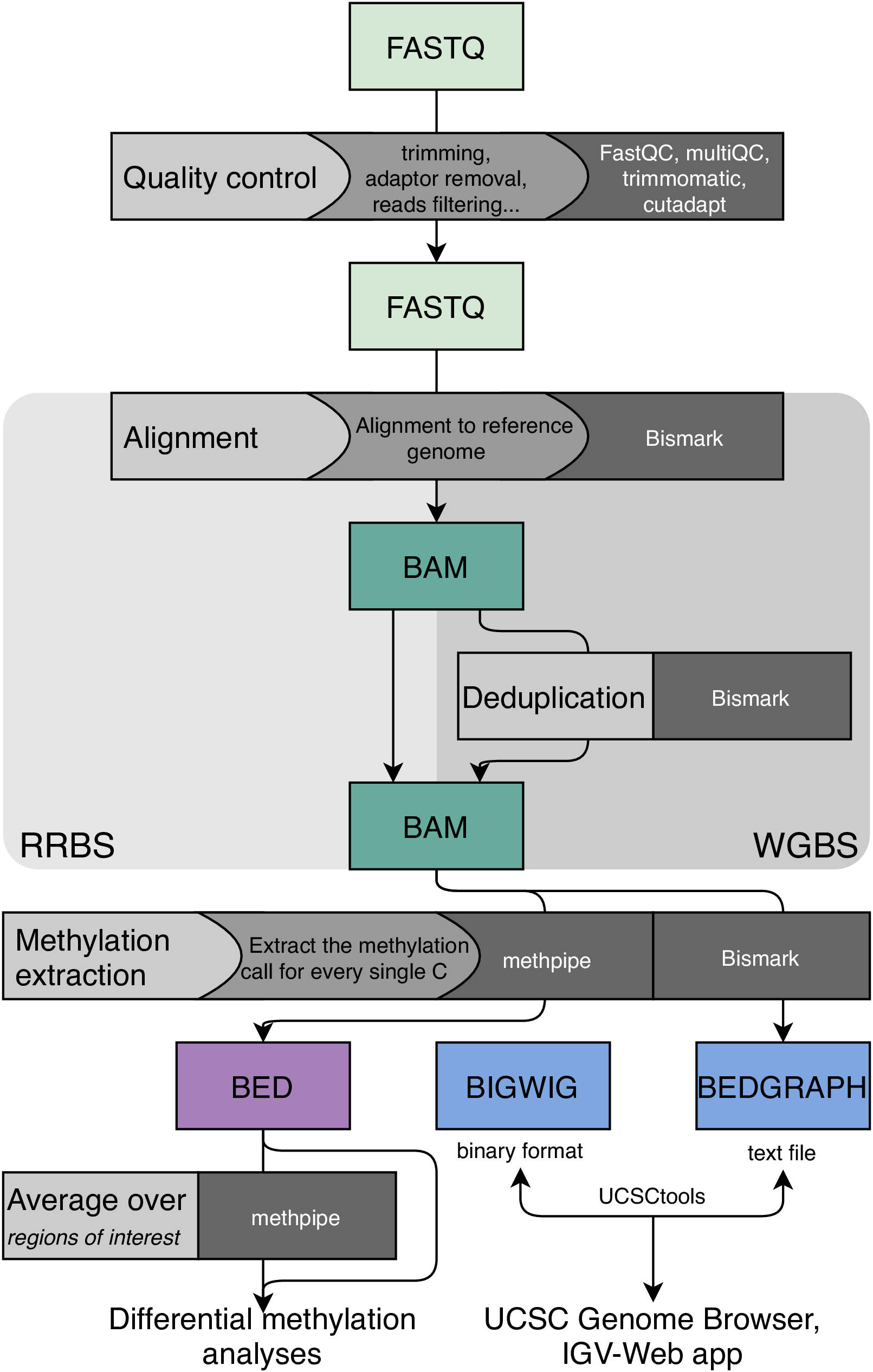
The objective of WGBS/RRBS data analysis is to identify the single nucleotide polymorphisms resulting from bisulfite conversion. Mapping of reads containing thymines, which potentially correspond to methylated cytosines and thus are no longer complementary to the reference genome, is challenging. The standard WGBS/RRBS processing pipeline implemented at the Core Bioinformatics group is shown in the scheme below. We perform WGBS/RRBS mapping to a reference genome using Bismark. Bismark estimates all possible C-to-T and G-to-A transformations in the reference genome before mapping each read. Mapped WGBS data is usually also subjected to deduplication to remove identical reads. Next, the levels of methylation at single-nucleotide resolution are extracted using the methpipe pipeline. The obtained methylation levels at single cytosines or across wider genomic regions can be further investigated via differential methylation analyses in order determine the differences between biological samples and/or conditions.